
Ateneos
LAS GRABACIONES DE LOS ATENEOS A PARTIR
DE 2020 SE ENCUENTRAN EN LA SOLAPA SOCIOS
Ateneos
Ateneo Hospital Garrahan. 16 de Julio del 2019
Ateneo tema “Gigantismo-Acromegalia”.Hospital de Pediatria Dr JP Garrahan.
El de julio se realizo en el aula virtual el ateneo sobre el tema Gigantismo-Acromegalia”.Las presentaciones fueron realizadas por las Dra Claudia Hernandez y la Dra Mirta Guitelman (Htal Durand).El ateneo fue coordinado por la Dra Isabel Di Palma (Hospital Garraham)
En forma presencial la audiencia fue de 80 profesionales y se conectaron de diferentes centros de nuestro país alrededor de cuarenta profesionales.
Paciente de 14 años de edad, sin antecedentes previos de relevancia, que consulta en agosto de 2018 por cefalea (de un año de evolución que en el último tiempo había incrementado su intensidad), mareos y visión borrosa de ojo derecho. Es evaluado por oftalmología y neurología quienes solicitan RMN cerebral que constata macroadenoma invasivo, por lo que se interna en Fundación Hospitalaria a fin de realizar cirugía descompresiva de manera urgente. Se realiza resección tumoral parcial, permaneciendo internado del 14 al 23/8/18. Se reciben dosajes hormonales basales dentro de parámetros normales. Por presentar agitación, mareos, tendencia al sueño, ptosis palpebral e hipofonía se reinterna del 27/8 al 21/9 interpretándose el cuadro como secundario a compromiso tumoral del tronco cerebral y probable insuficiencia adrenal, recibe tratamiento con anticonvulsivantes y dexametasona.
Se realizan los siguientes laboratorios (ver cuadro adjunto) donde se evidencia hiperactivación del eje somatotropo. Presenta además diabetes en tratamiento insulínico con bomba de infusión.
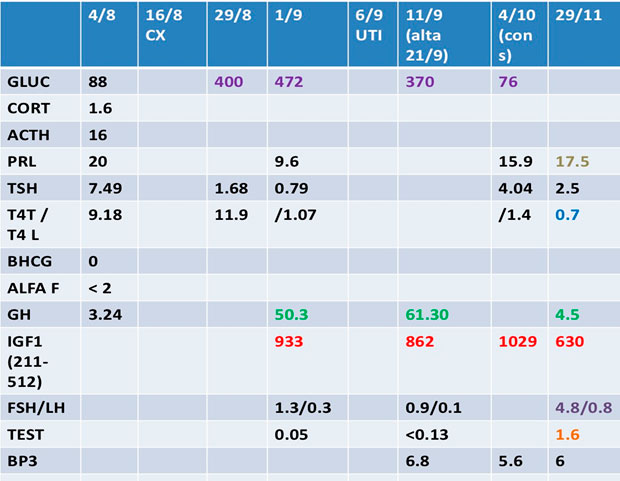
El estudio inmunohistoquímico arroja los siguientes resultados: GH (Somatotrofina) positiva citoplasmática en células dispersas. PRL (Prolactina): predominantemente negativa. Positividad citoplasmática en células aisladas. TSH (tirotropina) positiva citoplasmática en células dispersas. ACTH, FSH, LH: negativas. Ki 67: fracción de proliferación menor al 2-3%.
Con diagnóstico de Somatotropinoma se indica tratamiento con Análogo del receptor de somatostatina (SANDOSTATIN LAR) 30 mg/mes. Inicia el mismo el 4/10/18.
RMN SELAR realizada tras dos meses de tratamiento con SANDOSTATIN LAR destaca la presencia de voluminosa formación sólida con epicentro en región selar, señal algo heterogénea y moderado realce post contraste que se extiende en sentido lateral hacia ambos senos cavernosos englobando ambas carótidas. Presenta un gran componente latero selar derecho con efecto de masa y desplazamiento sobre la región temporomesial. Muestra además expansión nodular en sentido posterior y paramediana derecha la cual se proyecta hacia el ángulo pontocerebeloso ipsilateral. Diámetros máximos aproximados de la lesión de 33 x 55 x 62.
Se decide agregar al esquema terapéutico Cabergolina 1mg/ semana.
Desde el punto de vista clínico el paciente ha presentado mejoría, sin nuevos signos neurológicos, agudeza visual estable. Ha logrado reducir los requerimientos de insulina (hasta suspender en junio de este año).
Se encuentra puberal, estadio de Tanner 3. Datos antropométricos: P 79 kg (incremento de 14
kg). Talla 178 cm (por fuera de su rango genético).
Durante su evolución disminuyo la velocidad de crecimiento y retomo la progresión de su pubertad y continua con reemplazo hormonal con levotiroxina (100 mcg/día) e hidrocortisona (25 mg/día), además del tratamiento con octreotide(30 mg/mes) y cabergolina.(1 mg/semana)
El estudio molecular para el gen FIPA (11q13.3) fue realizado en el Hospital Italiano de BsAs por la Dra Lorena Viale, en el cual la secuencia fue normal, pero se hallaron tres variantes polimórficas que se presentan con mayor frecuencia en los pacientes con diagnóstico de FIPA (c.468 + 111 , c.682 C>A y c.920 A>G)
La bibliografía avala en estos pacientes la realización de estudios genéticos como parte de la evaluación del pronóstico de la enfermedad y respuesta al tratamiento ya que se ha visto que determinadas mutaciones se asocian a mala respuesta a drogas de primera línea requiriendo en esos casos rotar en el corto plazo la medicación dada la agresividad tumoral. Por otro lado, permitiría realizar el asesoramiento genético familiar y tomar conductas anticipatorias de detección precoz en el paciente y eventuales afectados ya que existen síndromes con asociación tumoral de distintos órganos.
Permanece bajo seguimiento multidisciplinario. Por el momento no se tomará nueva conducta neuroquirúrgica a la espera de respuesta favorable bajo tratamiento médico.
Ateneo Mendoza 7 de mayo 2019
Ateneo tema “Diabetes Neonatal”. Hospital de Pediatría Humberto Notti (Mendoza)
El de 7 mayo se realizó en el Hospital de Pediatría Humberto Notti de Mendoza el Ateneo ADEPA sobre Diabetes Neonatal. El mismo conto con la presentación del caso clínico por la Dra Carolina Colombi y las presentaciones a cargo de las Dras Sonia Peña (Htal Notti) y Patricia Taberner (Servicio de Nutrición y Diabetes. Hospital Elizalde (CABA))
Como discutidora de ADEPA participo la Dra Ana Vieites. Asistieron en forma presencial: 24 profesionales y se conectaron a través de internet alrededor de 90 profesionales de diferentes hospitales del país.
Caso clínico: Se presentó un paciente de sexo masculino, nacido a término, con peso adecuado para la edad gestacional, sin antecedentes perinatales relevantes. Segundo hijo de una pareja no consanguínea. Se lo recibió en el Servicio de Emergencias al mes de vida, en mal estado general, con dificultad respiratoria aguda y shock refractario a la infusión de fluídos.
Entre los resultados iniciales de laboratorio se constató glucemia de 5,11 g/L y acidosis grave con GAP aumentado. pH 6,7. pCO2 28. HCO3 3. Na 151 mEq/L. K 4,98 mEq/L. Cetonas positivas. Se inició aporte de insulina endovenosa continua. El requerimiento de insulina persistió por más de 15 días, por lo que se asumió que se trataba de una Diabetes neonatal.
Durante la internación manifestó diarrea profusa. Se descartaron causas infecciosas. Por la duración y severidad del cuadro, se sospechó enteropatía. Se agregaron eczema en rodillas e infecciones a repetición (por Pseudomona aeruginosa, Staphylococo aureus), Ig E elevada.
La asociación entre diabetes neonatal, enteropatía, eczema e infecciones recurrentes condujo al diagnóstico de Síndrome IPEX (Inmunodesregulación, Poliendocrinopatía, Enteropatía, ligado al X). Se realizó secuenciación Sanger del gen FOXP3 en Exeter (Reino Unido), resultando en el hallazgo de una variante nueva, hemicigótica, probablemente patogénica, en el exón 12. c.1282dup. p.(Thr428fs). La consecuencia es la pérdida del codón stop con la adición de 31 aminoácidos extra en el extremo C-terminal de la proteína FOXP3.
El síndrome IPEX se caracteriza por disregulación inmune, poliendocrinopatía, enteropatía, de herencia ligada al X. Es una enfermedad monogénica caracterizada por autoinmunidad multisistémica de inicio temprano, que puede comprometer la vida. Es un desorden hereditario raro causado por mutaciones con pérdida de función en el gen que codifica el factor de transcripción forkhead box P3 (FOXP3), que juega un rol clave en la diferenciación y función de las células T reguladoras naturales CD4+ CD25+, células esenciales para el establecimiento y mantenimiento de la tolerancia natural.
La diabetes neonatal se define como hiperglucemia que comienza en los primeros 6 meses de vida y requiere insulina para su tratamiento durante por lo menos 15 días. De acuerdo con el mecanismo etiológico se pueden clasificar en 3 grupos:
• Diabetes mellitus neonatal (DMN) causadas por genes que reducen el desarrollo pancreático (HNF1B, PDX1, PTF1A, GLIS3, NEUROD1, NEUROG3, RFX6, PAX6).
• DMN causadas por genes de producen destrucción de la célula beta (FOXP3, EIF2AK3, INS). Este grupo provoca diabetes neonatales permanentes.
• DMN causadas por genes que reducen la función de la célula beta (KCNJ11, ABCC8, GCK, SLC2A2, entre otros). Los pacientes que presentan mutaciones en KCNJ11 o ABCC8 pueden ser tratados con sulfonilureas a altas dosis.
El único tratamiento posible para la diabetes del IPEX es la insulina. El transplante de médula ósea está indicado en el Síndrome IPEX para tratar la desregulación inmune y la enteropatía.
Síndrome de IPEX. Dra Sonia Peña
Se reportó por primera vez en 1982 una familia con varones fallecidos en los primeros años de vida con clínica de diarrea, anemia hemolítica, diabetes y eczema, se presumió una herencia ligada al X y una sobreestimulación del sistema inmune. En 2001 se encontró la mutación en el gen responsable de la enfermedad, FOXP3. Este gen se localiza en la región centromérica de cromosoma X (Xq11.3–q13.3) y codifica una proteína FOXP3 de 431 aaaa, miembro de la familia FKH de factores de transcripción. Se han descripto 70 mutaciones. Las mutaciones con cambio de sentido y con cambio del marco de lectura que producen una proteína FOXP3 disfuncional se asocian a una clínica más severa. No hay una correlación estricta entre genotipo y fenotipo ya que lesiones en órganos target difieren en pacientes con la misma mutación.
Esta proteína FOXP3 se expresa en el núcleo de una clase de células T reguladoras (Tregs) que en su superficie expresan marcadores específicos como CD4, CD25 y CTLA4. La falta de expresión de la proteína FOXP3 o su inhabilidad de unión a DNA o a otras proteínas afecta el desarrollo y función de Tregs. Éstas cumplen función de regulación y de supresión de respuestas inmunitarias e inflamatorias a través de la liberación de citocinas como IL10 y TGF-beta y por supresión directa a otras células a través de su marcador CTLA4. Lo anterior provoca un desbalance inmune con hiperproliferación de LT efectores y expansión de LB autoreactivos con aumento en la producción de IgE y de autoanticuerpos. La agresión inmune resultante se caracteriza por la presencia de células inflamatorias que infiltran órganos target como intestino, páncreas, piel y tiroides, y la producción de autoanticuerpos contra intestino como HAAs (antiharmonin autoantibodies) y VAAs (anti-villin autoantibodies), contra el páncreas como GAD (antiglutamic acid decarboxylase), ICA (islet cell antibodies), IAA (insulin antibodies), IA2 (islet antigen2 antibodies), ZNT8 (zinc transporter 8) y contra tiroides TPO (anti-thyreoperoxidase antibodies) y TGB (anti-thyreoglobulin antibodies) y contra la piel anti- Keratine 8.
Diagnóstico:
Historia familiar confirmada o no de pacientes afectados varones en la rama materna. Historia de de Hidrops fetales y pérdidas fetales.
Fenotipo IPEX:
Diarrea acuosa/mucoide/sanguinolenta con desmejoría si se agregan fórmulas de inicio y que no responde a dietas de exclusión ni a ayuno.
Dermatitis presente o no desde el inicio eczematiforme, ictiosiforme, psoriasiforme o su combinación. Leve a severa.
DBT tipo 1 que precede o sigue a la enteritis. Destrucción del páncreas e infiltrado linfocitario intenso usualmente dirigido a los islotes pero también puede dañar tejido pancreático exócrino. Autoacs presentes o no. Cualquier endocrinopatía autoinmune en varones neonatos o pequeños debe tener seguimiento inmunológico para monitoreo de alguna otra manifestación autoinmune.
Alto riesgo de infecciones por disrrupción de barreras mucosas (piel, intestino) y por tto de inmunosupresión.
Enfermedad pulmonar intersticial respondedora a corticoides poco frecuente.
Severas alergias alimentarias aún en ausencia de endocrinopatías u otros síntomas autoinmunes.
Hallazgos de laboratorio:
Laboratorio con alteración de estado nutricional y metabolismo de glucosa. Hipereosinofilia usualmente. Igs normales salvo IgE ↑. Subpoblaciones linfocitarias normales. Células Tregs CD4+ CD25+ FOXP3 presentes o disminuidas. Expresión de proteína FOXP3 por citometría disminuida o ausente. Respuesta proliferativa in vitro normal.
Autoacs específicos detectables pueden predecir daño manifiesto en órganos target. Presencia de Acs anti-enterocito HAAs y VAAs. HAAs es específico de IPEX.
En neonatos con DBT y autoacs detectables IAA, ICA, GAD, o ZNT8, la búsqueda de mutación FOXP3 es mandatoria.
Tratamiento:
Trasplante alogénico con células progenitoras hematopoyéticas es curativo, mejor pronóstico a temprana edad y estadios tempranos de la enfermedad.
Casos con enteropatía severa , apoyo nutricional y terapia inmunosupresora (IS) precoz. Casos con endocrinopatía terapia de reemplazo con insulina/hormonas tiroideas. Citopenias autoinmunes, reemplazo con Hemocomponentes filtrados e irradiados y valorar Rituximab.
Profilaxis antibiótica/GGEV sobre todo ante uso de IS.
Corticoterapia sola o combinados con drogas inmunosupresoras para limitar la inflamación, pero no detiene la evolución de la enfermedad ni el inicio de nuevas manifestaciones. La inmunosupresión está dirigida a evitar la proliferación de LT efectores. Inhibidores de calcineurina (ciclosporina y tacrolimus) mayor toxicidad y menor especificidad afectando función de LT citotóxicos como Tregs. Inhibidores de vía mTOR como rapamicina (Sirolimus) actúa inhibiendo selectivamente la activación y en menor medida la proliferación de LT efectores, dando menor toxicidad.
Diabetes neonatal. Dra Patricia Taberner.
Si bien existían reportes sobre diabetes neonatal hace mas de 150 años, esta toma relevancia en el siglo XXI a partir de la observación de Diabetes Mellitus Neonatal (DMN) en un modelo animal transgénico que sobre expresaba el canal del K ATP.
En el año 2004 comienzan a dilucidarse las principales causas genéticas, es a partir de aquí que se definen sus características clínicas y pronósticas, así como su tratamiento apropiado.
La DMN es un síndrome heterogéneo monogénico raro cuya incidencia aproximada es un caso por cada 220.000 nacimientos. . Se la define como la hiperglucemia que comienza en los primeros seis meses de vida y dura por lo menos 15 días.
Los pacientes se caracterizan por ser RNTBPEG como consecuencia de haber sufrido retraso de crecimiento intrauterino, el cual es directamente proporcional a la deficiencia de insulina intrauterina, lo que confirma su importancia como factor de crecimiento durante el tercer trimestre de embarazo.
La DMN clínicamente se presenta en hiperglucemia aguda, cetosis o cetoacidosis, poliuria, deshidratación, ausencia de autoinmunidad beta detectable y péptido C bajo o indetectable.
La DMN es de naturaleza no autoinmune en la mayoría de los casos a excepción del síndrome IPEX (desregulación inmune, poliendocrinopatía entesopática ligada al X).
Se las clasifica en diabetes neonatal permanente (DMNP) y diabetes neonatal transitoria (DMNT), la primera comienza y continúa toda la vida, la segunda desaparece en semanas o meses y suelen recaer el 50% en la adolescencia.
También se las puede clasificar de acuerdo con el mecanismo etiológico en tres grandes grupos:
DMN causadas por genes que reducen el desarrollo pancreático (HNF1, PDX1, PTF1A, GLIS3, NEUROD1, NEUROG3, RFX6, PAX6)
DMN causadas por genes que producen destrucción de la célula B (FOXP3, EIF2AK3, INS)
DMN causadas por genes que reducen la función de la célula en la célula beta (KCNJ11, ABCC8, GCK, SLC2A2 entre otros).
La DMNP representa el 50% de los casos de DMN, las causas más importantes son las mutaciones activadoras en el gen KCNJ11, seguida de mutaciones en el gen de la insulina (INS), y en ABCC8.
Múltiples estudios experimentales sugieren que la mutación más frecuente asociada a DMNP (R201H, en KCNJ11) interfiere en la unión del ATP con la subunidad Kir 6,2 del canal de K de la célula beta lo cual reduce el efecto inhibitorio del ATP sobre esta subunidad.
La mayoría de los pacientes con DMNP por mutaciones en el gen KCNJ11 presentan diabetes aislada, mientras que un 20% tiene compromiso neurológico debido a la presencia de canales de K mutados en tejidos como cerebro, músculo esquelético y nervios periféricos. (Síndrome de DEND).
Las alteraciones clínicas en la DMNP producidas por mutaciones ABCC8 son similares a las observadas en KCNJ11, la principal diferencia entre los dos genes es que el Síndrome de DEND es más frecuente por mutaciones KCNJ11.
Las mutaciones en el gen de la insulina originan una síntesis anormal de la estructura de preproinsulina o proinsulina que conduce al estrés del retículo endoplásmico y posterior apoptosis de la célula beta, lo cual determina la caída progresiva de la secreción de insulina. Suelen comenzar los primeros seis meses de vida pero una minoría puede debutar después de los seis meses de vida y antes del año.
La DMNT si bien comienza en el mismo período de la vida que DMNP , suele debutar más tempranamente, remite en semanas o meses y reaparece en el 50% de los casos en la adolescencia por la insulino resistencia característica de la etapa puberal.
Representa el 50% del total de las DMN.
La hiperglucemia de la DMNT es el resultado de la reducción o ausencia en la producción de insulina en la etapa fetal que se extiende a la vida postnatal debido a un retraso en la maduración de los islotes pancreáticos y de las células beta como consecuencia de una alteración en la impronta genómica en el cromosoma 6q24, por trastornos de la metilación, isodisomía parental paterna o duplicación paterna, que afecta la expresión de los genes PLAG1 e HYMAI.
Otras causas de DMNT son las mutaciones activadoras en los genes ABCC8 y KCNJ11.
El diagnóstico se basa en la sospecha clínica y en la confirmación por medio del diagnóstico molecular por secuenciación del gen sospechado o por análisis de metilación, deleción/duplicación o disomía uniparental.
El tratamiento inicial es la alimentación adecuada y la insulinoterapia hasta la obtención del diagnóstico molecular. Una vez confirmado el diagnóstico, los pacientes que presentan mutaciones en gen KCNJ11 y ABCC8 pueden ser transferidos a sulfonilureas altas dosis. La droga de elección es la glibenclamida a altas dosis. Se hallaron pocos efectos adversos como diarrea, hipoglucemia, cambios de coloración en el esmalte dentario, alteraciones hematológicas o exantemas cutáneos.
Como conclusión los avances alcanzados en el campo de la DMN, gracias a la biología molecular condujeron a un inesperado cambio en el tratamiento de los niños que sufren esta enfermedad lo que les ha permitido desarrollar una mejor calidad de vida, a ellos como a su familia.
Ateneo ADEPA SAIC-SAP. 7 de noviembre 2018
Ateneo ADEPA -SAIC. 7 de noviembre del 2018. Tema “Tratamiento de bocio fetal hipotiroideo in útero. Reporte de un caso”.
El del 2018 se realizó en el Centro de Docencia y Capacitación Dr Carlos Giannantonio de la SAP el ateneo conjunto ADEPA-SAIC-SAP .Coordino dicho ateneo el Dr Guillermo Alonso (SAP-HIBA) y participaron como disertantes además del Dr. Alonso ,la Dra. Yamila Fittipaldi (HIBA) y como discutidor el Dr Lucas Otaño (HIBA).Como disertante finalmente participo la Dra. Carina Rivolta (SAIC) la cual actualizo el tema de “Genética molecular del Bocio congénito”.
Asistieron veintitrés profesionales y se contactaron vía zoom aproximadamente veinte profesionales
Caso clínico
Se recibe una mujer de 35 años, primigesta, cursando embarazo de 31 semanas con polihidramnios y masa cervical compatible con bocio fetal. Por dicho hallazgo es derivada a nuestra institución.
Presenta antecedente de insuficiencia hipofisaria múltiple secundaria adenoma hipofisario (resecado 10 años antes), medicada con levotiroxina 75 ug/d e hidrocortisona 10 mg/d. Requirió tratamiento inductor de ovulación para la concepción.
Ecografías previas a la consulta:
● 13 semanas y 20 semanas (scan fetal) normales.
● 31 semanas: feto con crecimiento en pc 72, polihidramnios, masa cervical que rodea tráquea compatible con bocio fetal e hiperextensión cervical. Doppler fetal normal, FCF 136 latidos/min.
Con dichos hallazgos ecográficos se decide realizar cordocentesis a las 32 semanas: TSH
191.4 uU/mL, T4 1.9 ug/dl, T4 libre < 0.4 ng/dl, T3 0.4 ng/ml, Tiroglobulina 8674 ng/ml (sangre fetal).
Se corrobora la sospecha diagnóstica de hipotiroidismo fetal, y se realiza en el mismo procedimiento amniocentesis (TSH 1.1, T4 1.2, T4 libre <0.4, T3 0.7) y amnioinfusión de 400 ug de LT4 soluble.
En la semana 33 se realiza nueva cordocentesis observándose franca mejoría de parámetros bioquímicos: TSH 11.6, T4 6, T4L 1, T3 0.5, TG 3835, junto con disminución del bocio y aumento de FCF.
Recibió un total de 6 infusiones semanales a dosis de LT4 400 ug, sin complicaciones y con controles ecográficos a los 4 días luego de cada infusión, presentando FCF estables, disminución del bocio y resolución de polihidramnios.
Nacimiento: Cesárea electiva a las 38 semanas. Recién nacida vigorosa, PN 2940 g, talla 48 cm, perímetro cefálico 35 cm, examen físico normal, tiroides no palpable.
En sangre de cordón: TSH 10.4, T4L 1.1, T4 8.6, T3 0.72.
Ecografía tiroidea: Lóbulo derecho 31 x 7 x 10 mm, lóbulo izquierdo 24 x 7.7 x 13 mm. Rx de ambos miembros inferiores con hipoplasia de núcleo distal de fémur.
Inició tratamiento con LT4 a las 48 hs de vida con ajustes periódicos según peso y datos bioquímicos.
A los 20 meses: presenta talla en pc 75, perímetro cefálico en pc 50, ausencia de bocio, hitos madurativos normales. Recibe LT4 4 ug/kg/día.
Estudio molecular pendiente.
Discusión. Dr Guillermo Alonso
El bocio fetal es un problema poco frecuente que puede resultar en complicaciones tanto obstétricas (polihidramnios, obstrucción esofágica, distocias de parto) como neonatales (obstrucción de vía aérea, distress respiratorio). El diagnóstico se basa en el hallazgo ecográfico. Habitualmente la historia clínica y el patrón ecográfico pueden definir la funcionalidad y las determinaciones hormonales en el feto rara mente son necesarias. Además deben considerarse otros diagnósticos diferenciales de masas cervicales y eventualmente el uso de resonancia nuclear magnética puede ser necesario.
Entre las causas de bocio fetal algunas pueden presentarse con hipertiroidismo (secundario a pasaje transplacentario de anticuerpos estimulantes del receptor de TSH (TSHr) en madres con antecedentes de enfermedad de Graves; mutaciones activantes del TSHr) o con hipotiroidismo (pasaje transplacentario de drogas antitiroideas o anticuerpos bloqueadores de TSHr; sobrecarga o deficiencia de Iodo materno; dishormonogénesis).
Presentamos el tratamiento con levotiroxina intramniótica en un feto con hipotiroidismo y bocio no autoinmune.
El uso de levotiroxina (LT4) por administración intrauterina en fetos presuntamente hipotiroideos fue utilizado inicialmente en el tratamiento de dos fetos presuntamente hipotiroideos por administración de iodo radioactivo como tratamiento de cáncer de tiroides a sus madres (Van Herle 1975; Lightner 1977).
Revisamos la literatura y encontramos descripciones del uso de LT4 como tratamiento de bocio fetal en 38 pacientes. El 1er reporte es de Perelman, 1990. Un estudio francés (Rivault 2009) muestra los datos en una serie retrospectiva de 12 pacientes. El resto son casos aislados y revisión de literatura.
La edad gestacional al diagnóstico ecográfico de bocio se realizó a una edad mediana de 25 semanas con un rango entre las 18 y 34 semanas de edad gestacional. En la mayoría de los casos, el diagnóstico fué realizado en el contexto de la ecografía de control que se realiza habitualmente entre la 22 y 24 semanas. Además del hallazgo de bocio otros datos descriptos son polihidramnios (en 20 de 25 reportes), hiperextensión cervical, disminución del contenido gástrico, disminución de la frecuencia cardíaca fetal o retraso en la maduración esquelética.
En el 48% de los pacientes se utilizó la sangre fetal (SF) obtenida por cordocentesis para certificar el diagnóstico de hipotiroidismo. En 21% (8 pacientes) se tomó muestra simultánea de líquido amniótico (LA) para dosajes hormonales. En 26% el diagnóstico bioquímico se basó en el dosaje en LA exclusivamente. En dos pacientes el diagnóstico de hipotiroidismo se justificó sólo en datos clínicos.
El dosaje en sangre fetal de TSH al diagnóstico presentaba una mediana de 98.5 mU/L con un rango entre 1.9 y 472. La TSH en LA fue 2.1 mU/L (mediana) con un rango entre 0.02 y 99.5. No se encontró correlación entre los dosajes en SF y LA en los 8 pacientes en que se tomaron muestras simultáneas.
La primera dosis de LT4 intramniótica se administró a una edad gestacional mediana de 30 semanas repitiéndose dosis con intervalos variables. Los pacientes recibieron entre 1 y 11 dosis (mediana 3) y las dosis también fueron variables (entre 100 y 500 ug/dosis). La mediana de dosis total recibida fue 950 ug.
La etiología del hipotiroidismo fue asumida como dishormonogénesis confirmada por biología molecular en 4 pacientes (3 deficiencia de TPO, 1 deficiencia de DUOXA2), pasaje transplacentario de anticuerpos (1 paciente), sobrecarga de iodo (1 paciente) y pasaje transplacentario de drogas antitiroideas (5 pacientes). Se presume hipotiroidismo por dishormonogénesis en los restantes 27 pacientes.
En cuanto a la evolución, la literatura refiere disminución del tamaño del bocio fetal en 27 de 30 pacientes así como disminución de polihidramnios. En 4 pacientes el parto se produjo antes de las 37 semanas. Se refiere nacimiento por parto vaginal en 17 embarazos y por cesárea en 9.
En relación a los recién nacidos, se constató la presencia de bocio en ⅔ e ellos, con una media de TSH al nacer de 99mU/L (rango 0.04-450). Nueve de estos pacientes tenían TSH menor a 20 mU/L. En sólo 6 pacientes se refieren datos respecto a radiología del nucleo de Beclard siendo anormal en 5 de ellos. Ninguno de los pacientes requirió maniobras para preservar permeabilidad de la vía aérea. La neuromaduración es descripta como normal en 19 de 19 pacientes reportados.
En estos 38 casos se refiere, como única complicación, un caso de corioamnionitis que precipita el parto a las 32 semanas. Por otra parte, encontramos un reporte reciente de muerte intrauterina a las 31 semanas en un feto con bocio luego de la segunda infusión amniótica (Vasudevan 2017).
En resumen, presentamos el tratamiento exitoso en forma intrauterina de una niña con hipotiroidismo congénito y bocio. El mismo se realizó con infusiones semanales de LT4 en un centro de alta complejidad y con experiencia en medicina fetal.
Reporte Ateneo conjunto ADEPA
Reporte Ateneo conjunto ADEPA – Hospital Universitario La Paz Madrid
Fecha: 7 de junio 2018
Tema: “Infancia y adolescencia trans: el rol del Endocrinólogo Pediatra”.
Presentaciones: Dra Verónica Fernández Mentaberry (Hospital Durand), Dra Verónica Figueroa (Hospital Elizalde), Dra Cristina Mora (Hospital La Paz), Dr Julio Guerrero Fernández (Hospital La Paz)
La presentación de los expositores estuvo a cargo de la Dra. Claudia Hernández. En primer lugar, realizó una breve introducción la Dra. Isabel González Casado, Jefe del Servicio de Endocrinología del Hospital La Paz. Luego, las Dras Verónica Fernández Mentaberry y Verónica Figueroa realizaron primeramente la presentación de dos pacientes y una aproximación general acerca de la temática Trans y posteriormente se refirieron a la experiencia conjunta de ambos hospitales.
La Dra. Cristina Mora Palma se refirió al tema “Terapia de reemplazo en los pacientes trans” describiendo las características de la población tratada y las modalidades de tratamiento empleadas en su tratamiento. El Dr Julio Guerrero Fernández se refirió a los efectos adversos asociados a la terapia hormonal.
Esta temática permitió un rico intercambio entre los disertantes y los asistentes. Dado lo novedoso de esta problemática en edad pediátrica y durante la adolescencia, se generaron nuevos interrogantes.
Quedan pendientes nuevos estudios prospectivos y un seguimiento más prolongado que permitan responder algunos de estos interrogantes de forma más categórica.
Para esta actividad, se contaron profesionales desde 14 centros de nuestro país, otros lo hicieron a través de redes domiciliarias en nuestro país y en el exterior, alcanzándose un número aproximado de 172 profesionales médicos (pediatras, endocrinólogos, hebiatras) bioquímicos y psicólogos.
JORNADA ADEPA DE ENDOCRINOLOGIA PEDIATRICA
Jornada ADEPA Endocrinología Pediátrica, 06 de octubre de 2017, Salta.
Caso Clínico: HIPOCALCEMIA
Dr. Julio F. Nader. Hospital Público Materno Infantil, Salta.
Edad 1° consulta: 13 años, Motivo consulta: evaluación tiroidea, hipocalcemia
Enf. actual: Enfermedad de Hodgkin estadio IIa de reciente diagnóstico. Antec. Enf. actual: consulta en 8/2014 por tumoración cervical, por punción biopsia se hace Dco. Enf. Hodgkin en 9/2014. Se hace centellograma con citrato de Ga 67 + TAC cuello/tórax/abdomen para estadificación, y por compromiso de varios grupos ganglionares + localización supradiafragmática + ausencia de síntomas generales se asume: Enfermedad de Hodgkin estadio IIa.
Laboratorio: inicial pre inicio Qcx (9/14): Ca 9,2 mg/dl y P 4,6 mg/dl (normal). Se decide iniciar Qcx protocolo ABDx (son 4 drogas) consistente en 6 ciclos (2 bloques cada ciclo) en 11/2014. Hace 1° bloque del 1° ciclo y el 1° día del 2° bloque se recibe laboratorio con Calcio bajo y fosforo elevado. Se asume probable síndrome lisis tumoral por laboratorio, se suspende Qcx e inicia hiperhidratación + calcio EV sin respuesta favorable.
Por baja asociación de lisis tumoral en Enf. de Hodgkin (masa tumoral pequeña) + falta de respuesta a hiperhidratación asociado a Calcio EV + hipocalcemia persistente + PTH baja 4,68 pg/ml se asume: Hipoparatiroidismo primario por 1) infiltración tumoral glandular, 2) compromiso vascular por tumor, 2) daño por quimioterapia.
Se inicia tto con Calcitriol + Calcio vía oral con buena evolución clínica. Se reinicia Qcx días después de normalización calcemia y completa los 6 ciclos con final en 6/2015. Al finalizar Qcx (6 meses después dx) se intenta suspender Calcitriol con caída niveles de Calcio y elevación fosforo asumiendo: Hipoparatiroidismo primario permanente. Dos 2 años y 9 meses después: Enfermedad Hodgkin en remisión, hipoparatiroidismo 1° permanente. Tratamiento: Calcitriol 3ug/d (12 capsulas/día) + Calcio 2 g/día.
HIPOCALCEMIA: DIAGNÓSTICO Y SU MANEJO
Dr. Hamilton Cassinelli. Médico Pediatra Endocrinólogo. División de Endocrinología, Hospital de Niños Ricardo Gutiérrez
Hipocalcemia se define como la disminución de las concentraciones de calcio total por debajo de la cifra de referencia, que habitualmente es de 8,5 mg/ dl. Para evitar dudas podemos recurrir a las cifras de calcio iónico, que se consideran de hipocalcemia cuando son inferiores a 4,6 mg/dl. Hay que diferenciar de la Hipocalcemia Ficticia: hipocalcemia por hipoalbuminemia, cuando albúmina cae <4.0 g/dl, la corrección es sumar 0.8 mg/dl calcio por cada 1.0 g/dl albúmina disminuida.
Características Clínicas Asociadas con Hipocalcemia Neuromuscular: Signo Chvostek, Signo Trousseau, Parestesias, Tetania, Convulsiones, Calambres Musculares, Espasmo de Laringe, Espasmo Bronquial, y Prolongación intervalo QT en ECG.
HIPOPARATIROIDISMO: Grupo de enfermedades originadas por secreción deficiente o disminución de la acción periférica de la Paratohormona (PTH) llevando secundariamente a un cuadro consistente en hipocalcemia e hiperfosforemia. Puede ser congénito o adquirido, transitorio o permanente.
Hipoparatiroidismo Neonatal:
- 24-48 horas de vida hipocalcemia neonatal temprana puede ocurrir y es más común en niños prematuros, niños hijos de madres diabéticas, niños con SDR
- hipocalcemia neonatal tardía 5-10 días de vida, niños a término, hiperfosfatemia por administración leche de vaca o alimentos fosfatados, Deficiencia Magnesio, Hiperparatiroidismo materno durante el embarazo.
HIPOPARATIROIDISMO. Clasificación:
A) CONGÉNITO
Neonatal Transitorio
Familiar Aislado: Mutaciones en gen de PTH: (defectos en la síntesis) AD o AR, Mutaciones activantes en CASR: (defectos en secreción de PTH) AD, Mutaciones en GCMB: AR, Hipoparatiroidismo Idiopático: AR o ligado al X.
Síndromes Multisistémicos Congénitos: Di George & Velocardiofacial (22q11), Barakat / HDR, Kenny-Caffey & Sanjad-Sakati.
Neuromiopatías mitocondriales (Síndrome de Kearns Sayre)
Resistencia a PTH: Pseudohipoparatiroidismo, Condrodisplasia de Blomstrand (mutación de PTHR1).
B) ADQUIRIDO: Autoinmune SPGA Tipo I (APS-1 o APECED), Postquirúrgico, Radiación, Infiltrativo.
Manejo de la hipocalcemia:
Gluconato de calcio intravenoso es el preferido, ya que cloruro de calcio causa irritación local. Gluconato de calcio contiene 90 mg calcio elemental por 10 ml, usualmente 1-2 ampollas (180 mg elemental calcio elemental) diluidas en 50-100 ml de dextrosa 5% infundida durante 10 minutos. El gol Standard es aumentar el calcio 2 a 3 mg/dl con la administración de 15 mg/kg del calcio elemental durante 4 a 6 horas. Calcio debe ser mantenido en el límite inferior normal. Si es posible, la suplementación oral con calcio concurrentemente con 1 a 2 gramos de calcio elemental y 1,25-dihidroxivitamina D.
Carbonato de calcio oral es el más comúnmente utilizado. Calcio en cantidad de 1 a 3 gramos de calcio elemental en 3-4 dosis divididas con las comidas asegura una óptima absorción. Carbonato de Calcio contiene 40% calcio elemental. Cantidades menores de calcio elemental están presentes en otros tipos de calcio: lactato de calcio (13%), citrato de (21%) y gluconato de calcio (9%), requiriendo mayores cantidades de tabletas.
CASO CLÍNICO CUSHING EXÓGENO
Mariangeles Insúa Beverina. Médica Pediatra Endocrinóloga. Hospital Público Materno Infantil, Salta
Paciente sexo masculino de 8 meses de edad derivado desde el hospital del lugar de origen por bajo peso y baja talla. MC: desnutrición, baja talla severa.
Presenta como antecedentes: 2º hijo de padres jóvenes, no consanguíneos, embarazo controlado, parto normal, PN: 2,600 Kg. TN: 47 cm, PC: 33 cm EG: 38 SEG. RNT/AEG. No internaciones en neonatología. Lactancia materna mixta desde el nacimiento hasta los 2 meses de vida, luego solo fórmula de inicio refiriendo la madre con regular tolerancia. Incorporación de leche entera a partir de los 7º meses.
Enfermedad Actual: Según se constata en el carnet de sus controles en lugar de origen, se evidencia evolución favorable de peso hasta el 3º mes, con peso estacionario desde el 4º mes hasta el momento de la primera consulta cuando presenta kg 5,100 con -4,49 DS Z SCORE. Con respecto al crecimiento en talla, se observa también descenso marcado en curva con caída en la velocidad de crecimiento a partir del 3º mes presentando al momento de la consulta talla de 56 cm - 6,76 DS en Z SCORE.
En el interrogatorio se constata que el niño presenta cuadros respiratorios a repetición con componente bronquial obstructivo desde los 2 meses de edad, recibiendo en casi todo cuadro catarral y/o bronquial de forma frecuente y prolongada (generalmente más de 1 semana x ciclo) indicación de tratamiento con corticoides exógenos (betametasona / dexametasona), habiendo sido su última dosis 15 días previos a la consulta luego de 15 días de tratamiento por un resfrío común.
Durante el exámen físico al momento de la consulta: Paciente en buen estado general, crónicamente enfermo, hidratado, funciones fisiológicas conservadas. Facie redondeada, bolas de bichat prominentes, plétora facial, tórax adelgazado, abdomen prominente distendido, miembros inferiores y superiores adelgazados con hipotrofia marcada a nivel de muslos. Desarrollo pautas madurativas: paciente con retardo en la adquisición de las pautas.
Se asume como Sd de Cushing exógeno por corticoides asociados a los cuadros respiratorios a repetición. Se indica evitar el uso de corticoides orales y/o IM en el tratamiento de los cuadros respiratorios (salvo necesidad) y mantener manejo sintomáticos de los mismos. Se deriva a neumonología para valorar realizar Test del sudor debido al antecedente de cuadros respiratorios obstructivos a repetición y mala ganancia pondoestatural (resulta muestra insuficiente).
Se cita a control a los 2 meses. En el control siguiente se evidencia en el exámen físico: Peso: 5,800 kg (aumentó 700 grs en meses), Talla:63,5 cm, creció 7,5 cm disminuyendo percentilo de - 6,76 a -4.27 DS Z SCORE. Talla sentado 38 cm Relación talla sentado/talla en P10. Se evidencia disminución marcada de la facie en luna llena, con disminución del tejido adiposo de las bolas de bichot, disminución de la plétora facial, mejoría en el tono y trofismo miembros sobre todo de miembros inferiores a nivel de muslos. Evolución: el mes previo presentó nuevo episodio respiratorio bronquial obstructivo con requerimiento de corticoides endovenosos nuevamente. No realizó tratamiento preventivo indicado por neumonología con budesonide (aerosolterapia). Se solicita fórmula con hidrolizado proteico por sospecha de APLV ante cuadros respiratorios a repetición, diarrea frecuente y mala ganancia pondoestatural. Se solicita valoración cardiológica: exámen cardiovascular norma. Se solicita valoración gastroenterología: se indica continuar con fórmula hidrolizada
EFECTOS DE LOS CORTICOIDES EN EL CRECIMIENTO Y MASA OSEA
Dra. Ana Carolina Arias Cáu. Servicio de Endocrinología, Hospital Materno Infantil Dr. Héctor Quintana, San Salvador de Jujuy.
Caso clínico: Paciente masculino de 13 años, consulta a la guardia de pediatría por vómitos incoercibles y dolor en zona lumbar. Refiere CVAS, disfonía y tos de 1 semana de evolución. Posterior a un acceso de tos refiere dolor intenso en zona lumbar que aumenta luego de querer pararse repentinamente. La madre lo medica con analgésicos y reposo. Hace 24 hs. inicia con vómitos (4 hasta el momento) y debilidad en miembros inferiores por lo cual consulta a la guardia de pediatría.
Antecedentes Enfermedad Actual: Tratamiento prolongado con Glucocorticoides desde hace 1 año y medio por Artritis Crónica. Actualmente recibe Meprednisona 8 mg/día, desde hace 1 mes, ya que está en descenso por mejoría clínica. Además, Calcio vía oral y vitamina D.
Es examinado por guardia, dolorido, nauseoso, pálido, sudoración superficial, lloroso. Facies cushingoide, giba dorsal blanda, hirsutismo, obesidad troncal. Estrías rojas más de 1 cm en abdomen, axilas y muslos. Abdomen blando, doloroso a la palpación sin defensa ni contractura. Debilidad muscular. Dolor a la compresión en zona dorso lumbar. Dificultad para sentarse. Taquicárdico y mal perfundido.
Es ingresado con diagnósticos de: 1) Intolerancia Gástrica con deshidratación, 2) Crisis adrenal gatillada por dolor, 3) Lumbalgia. Se colocó acceso venoso, Push de hidrocortisona EV 100 mg, hidratación y analgesia. Los laboratorios de la muestra crítica mostraron Cortisol ND y ACTH <10pg/ml, Na/K: 135/5.8 y el resto del laboratorio normal.
En la reevaluación el paciente mejoró francamente su condición hemodinámica y el abdomen blando, depresible e indoloro, pero agudizó el dolor lumbar a pesar de la analgesia. La Radiología de Columna mostró múltiples fracturas de columna dorsal y lumbar, por lo cual el paciente fue reevaluado por endocrinología. Continuó con aporte de calcio y vitamina D y a las 48 hs. reinició tratamiento con Meprednisona.
Examen físico: EC: 13 a y 6 meses, Peso: 55 kg (P75-90), Talla: 138.5 cm (P10), BMI: 28 gr/cm², FC 90 x min, TA: 80/65.Piel suave, eutérmica, no bocio, estigmas Cushing, Estrías, lipomastia, Tanner: G2, VP 3, testículos 5 ml. Edad ósea: 11 años. TOG: 174 +- 6 cm. Se asumió al paciente como osteoporosis secundaria a glucocorticoides y se analizó el metabolismo mineral óseo en donde se observó una marcada deficiencia de vitamina D (<5ng/ml) y se consideraron:
Dolor intolerable por fracturas vertebrales - Factores de riesgo para nuevas fracturas: 1) Hipercortisolismo prolongado, 2) Detención del crecimiento y desarrollo Puberal por enfermedad de base más los GC, 3) Obesidad mórbida, 4) Inmovilización.
Conducta: Se indicó infusión con Pamidronato dosis de 30 mg/dosis sin efectos adversos con los objetivos de calmar el dolor y prevención de nuevas fracturas.
Conclusiones: Tanto la actividad de la enfermedad de base del paciente como la exposición prolongada a glucocorticoides (GC) manifestaron efectos en diferentes sistemas: fracturas vertebrales secundarias a osteoporosis por GC, crisis adrenal gatillada por dolor y deprivación de GC, detención del crecimiento y de la pubertad.
La sospecha clínica y tratamiento de la crisis adrenal en urgencias permitió compensar al paciente y evitar serias consecuencias. El tratamiento del dolor y la reevaluación fueron de suma importancia para los diagnósticos diferenciales. El tratamiento con Bifosfonatos ayudó en la resolución aguda del dolor y permitió a largo plazo que el paciente pudiera mejorar su masa ósea. El suplemento de calcio y vitamina D fueron importantes suplementos durante la evolución de la enfermedad para la preservación del hueso.
Comentario final: Los Glucocorticoides en exceso son perjudiciales para el crecimiento y el desarrollo. El hipercortisolismo exógeno es muy frecuente en niños y adolescentes; y las complicaciones son: el síndrome de Cushing y la inhibición del eje adrenal. El impacto negativo en la masa ósea tanto en corto y largo plazo, sobre todo durante los primeros años de vida y la pubertad, aumentan considerablemente el riesgo de fracturas en la vida adulta.
La asistencia interdisciplinaria de estos pacientes es una herramienta indispensable para prevenir y evitar complicaciones.
Es importante el acompañamiento por el endocrinólogo pediatra para aquellos pacientes con corticoterapia prolongada, para brindarles medidas de prevención. Las mismas incluyen incentivar la ingesta de lácteos y alimentos ricos en calcio, el aporte de calcio vía oral según los requerimientos por edad y vitamina D al doble o triple de la dosis requerida según la edad del paciente.
PAUTAS ANTIESTRES
Dra. Claudia Hernández, Medica Pediatra Endocrinóloga, Hospital de Niños Pedro de Elizalde. CABA.
Los glucocorticoides exógenos son moléculas similares al cortisol, a los cuales se le realizan modificaciones químicas que le confieren características farmacológicas específicas como:
Efecto mineralo y glucocorticoide /potencia / acción antiinflamatoria / duración de acción
Su uso a dosis farmacológicas en pediatría es muy frecuente, siendo la dexametasona, betametasona y prednisona los más utilizados. Las enfermedades respiratorias agudas son la primera causa de su uso, utilizándose ciclos cortos pero repetidos en el tiempo, también son utilizados en periodos más largos en enfermedades crónicas de origen reumático, dermatológico, nefrológico, oftalmológico, etc.
El eje Hipotalamo-Hipofiso-Suprarrenal es susceptible de ser inhibido cuando el paciente se encuentra en tratamiento con glucocorticoides. Debido a ello, se requieren un aumento de la dosis de corticoides exógenos en situaciones de estrés, para evitar que se desencadene una crisis adrenal; dado que el eje adrenal en condiciones normales se activa durante episodios de estrés, presentando valores de cortisol entre 40-50 ug/dl. La terapia crónica con glucocorticoides (GC) está asociada a múltiples efectos adversos con alto impacto en la calidad de vida, por lo que su uso debe ser racional, utilizando de ser posible:
- La menor dosis necesaria/el menor tiempo posible/una dosis al día/dosis en días alternos
Todo paciente con corticoterapia prolongada se considera insuficiente adrenal, y esta corticoterapia prolongada es la primera causa de insuficiencia suprarrenal en niños y adultos
- El paciente y sus familias deben ser educados ante una emergencia
Se debe indicar pautas antiestrés a todo paciente que:
• Haya recibido GC en forma prolongada y su función adrenal no haya sido evaluada.
• Se encuentre en plan de descenso de GC
• Presente insuficiencia suprarrenal establecida
• Como profilaxis de cirugía de tumores selares y paraselares
• Aquellos que hayan recibido medicación que interfiera en el metabolismo del cortisol
- Estas deben ser individualizadas y actualizadas periódicamente
El paciente y sus familias deben ser educados ante una emergencia.
|
LEVES – MODERADAS
|
GRAVES
|
CIRUGIA
|
|
Fiebre, dolor, procesos dentales, infecciones leves sin vómitos (buen estado general).
|
Cirugías, infecciones severas |
Programada o de urgencia profilaxis
|
|
Duplicar dosis de Hidrocortisona por vía oral, durante la duración del proceso (48-72 hs)
Intolerancia oral Hidrocortisona 30 mg/m2/dosis EV o IM Hasta tolerancia oral Luego seguir con doble dosis Vía oral hasta la recuperación
|
Hidrocortisona 60 mg/m2/dosis, EV, en bolo. Seguir con 60 mg/m2/día cada 4 a 6 hs o en goteo continuo durante 48 hs. Continuar como estrés leve por 48 hs antes de volver a la dosis habitual
|
Hidrocortisona 1 hora antes • 60 mg/m2/dosis durante la cirugía. • 60 mg/m2/dosis Luego continuar • 60 mg/m2/día cada 4 a 6 hs durante 24-48 hs. Luego volver a la dosis habitual
|
| Cuando se administra ev se fracciona cada 6 horas
Cuando se administra im se fracciona cada 8 horas |
||
| Dosis por peso corporal
Hasta 10 kg 25 mg De 10 a 30 kg 50 mg/día Mayor 30 kg 80-100 mg /día |
||
Ateneo ADEPA a Distancia 2017
ATENEO A DISTANCIA 2017
Realizado por la Sección Crecimiento, Desarrollo y Endocrinología. Servicio de Pediatría. Hospital Nacional. Prof. A. Posadas. El martes 22 de agosto de 2017, 9.30hs. desde la Oficina de Comunicación a Distancia (OCD) del mismo Htal.
Se conectaron desde diversos lugares del país, CABA: Hospital Nacional de Pediatría JP Garrahan (28 participantes), Hospital Italiano (5 part), Hospital de Niños Ricardo Gutiérrez (35part), Pcia de Buenos Aires, La Plata: Hospital de Niños Sor Maria Ludovica (7 part), Hospital Alejandro Posadas (17 part), Salta: Hospital Materno Infantil (2 part), Hospital Castro Rendon Neuquén (5 part), Hospital de Niños de Córdoba (8 part), Tucumán: Hospital Nicolas Avellaneda (7 part), Jujuy: Hospital Quintana (2 part), y Bahía Blanca (sin registro), Total: 116 participantes.
Caso Clínico: Hipoparatiroidismo. Uso de rhPTH
Paciente de 14.7 años, de sexo femenino derivada con diagnóstico de Hipertiroidismo de un año de evolución, bajo tratamiento con Metimazol 50 mg/d y Propanolol 120 mg/d con cumplimiento irregular. Ingresó en regular estado general, con bocio (tiroides 80 g, aumentada de consistencia, superficie abollonada), exoftalmos leve, obesa, con cuadro compatible con crisis tirotóxica (Criterios Burch y Wartofsky:50), enfermedad de Graves-Basedow. Se realizaron exámenes de rutina normales, Anticuerpos celiaquía negativos, T4L: >7,77 ng/dl (vr: 0.65 -2.0), T4: > 24,8 ug/dl (vr: 6.09 -12.23 ) TSH: 0,01mUI/ml (vr: 0.49-4); TRAb: 73%, (vr: hasta 15) ATG y ATPO: negativos Ca: 10.3mg/dl (vr: 8.6-10.2) P:4.0 mg/dl (vr: 2.5-4.5); Mg: 2.0 mg/dl (vr: 1.6-2.6); FAL: 147 U/L (vr: 35-105); PTH:28 pg/mL (vr: 12-88); Vitamina D: 23 ng/ml. Ecografía tiroidea: ligeramente heterogénea con vascularización aumentada en forma simétrica LD: 66x25x33 mm, LI: 60×23 x24mm, istmo 5 mm. Se aumentó Metimazol hasta 120 mg/d y Propanolol 240 mg/d, sin respuesta al tratamiento médico se realizó tiroidectomía total. Anatomía Patológica: carcinoma papilar bilateral multifocal variante clásica diámetro (> 0.9 cm), un ganglio linfático libre de infiltración neoplásica y 2 glándulas paratiroideas. Estadío: T1a, N0, Mx
Laboratorio postquirúrgico (3 hs) Ca: 9.1mg/dl; P: 3.3 mg/dl, Ca iónico: 1.26 pmol/l, Mg: 1.8 mg/dl, PTH: < 10 pg/ml.
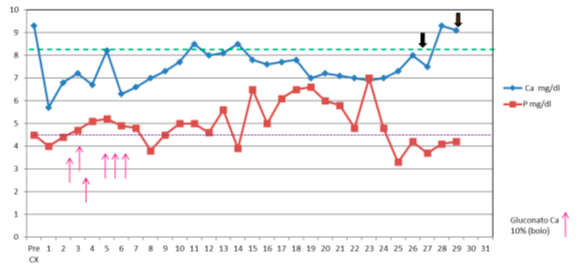
24 hs post quirúrgico presentó hipocalcemia sintomática (gráfico 1) (calambres y parestesias peribucales) Ca T: 5.7 mg/dl, Ca i: 0.84 pmol/l, P: 4.7 mg/dl, Mg: 1.8 mg/dl, Ca/creat: 0.02- 0.05. Requirió repetidos bolos de gluconato de Ca e.v, e infusión continua e.v hasta 2g/d y carbonato de calcio v.o hasta 6 g/d. La dosis de calcitriol fue gradualmente aumentada hasta 2 mcg/d y Sulfato de Mg 25 % e.v en infusión continua. Recibió Vitamina D 300.000 UI (50.000 UI c/15 días) Se interpretó como síndrome de hueso hambriento secundario a hipertiroidismo manifestándose con hipocalcemia severa y fósforo normal que se fue elevando a partir del día 15. Sin respuesta al tratamiento convencional se interpretó como hipoparatiroidismo postquirúrgico indicándose rhPTH (1-34) 20 ug/d vía s.c .12 hs post el Ca subió inmediatamente de 7.4mg/dl a 9.3mg/dl; P: 4.1 mg/dl Mg: 2.0 mg/dl, disminuyéndose el aporte de calcio y calcitriol. Ecografía renal: normal; DMO corporal total: Z Score -0.6, DMO columna lumbar L1-L4: Z Score -0.4. Requirió 2 internaciones a los 45 y 60 días post quirúrgica por suspensión de rhPTH por hipocalcemia sintomática recibiendo gluconato de calcio bolo 2 g/24 hs e.v en infusión. Se indicó rhPTH: Ca: 5.9 mg/dl, P: 8.4 mg/d, Mg: 1.8 mg/dl, post rhPTH: Ca: 9.1 mg/dl; P: 4.0 mg/dl; Mg: 1.9 mg/dl.
Dra. Silvia D’Amato: Médica Pediatra Endocrinóloga. Hospital Nacional Prof. A. Posadas.
Discusión:
El hipoparatiroidismo se define como la presencia de hipocalcemia e hiperfosfatemia con valores bajos o inadecuados de PTH intacta. Dentro de todas las causas el hipoparatiroidismo post quirúrgico es el más frecuente en nuestro medio, seguidas por las alteraciones del receptor sensible al calcio y las autoimunes. Su signo-sintomatología es variable de acuerdo con el tiempo de evolución de la hipocalcemia. Las complicaciones más temidas son secundarias al aumento del producto calcio/fósforo secundario y exacerbado por el tratamiento que el hipoparatiroidismo requiere como lo son las sales de calcio, la vitamina D activa y el colecalciferol. Puede producirse litiasis renal con hipercalciuria (se maneja con tiazidas), nefrocalcinosis, calcificaciones de ganglios de la base, convulsiones, depresión y trastornos cardiovasculares entre otros.
Desde el año 1996 se comenzaron los estudios con PTH 1-34 y la sustitución en el hipoparatiroidismo. Se realizaron múltiples estudios que compararon dar PTH 1-34 20 mcg/día de manera subcutánea Vs tratamiento con calcio, calcitriol y vitamina D. Es claro el beneficio de dar PTH 1-34 para bajar los niveles de calciuria, manteniendo los valores de calcemia normales, estimulando la excreción de fósforo urinario (por su acción fosfatúrica). Produce aumento de los marcadores de remodelado óseo (cross laps, Fal ósea) pero al día de la fecha los estudios no son contundentes para demostrar impacto en la masa ósea. En el hipoparatiroidismo los marcadores de remodelado óseo se caracterizan por estar planos con una densidad ósea conservada, con aumento del volumen óseo con mayor densidad y número de trabéculas, se necesita esclarecer si dichas características de la microarquitectura ósea predisponen a fragilidad ósea y mayor riesgo de fracturas. Dentro de los efectos adversos descriptos al administrar la PTH se encuentran naúseas, vómitos, hipotensión arterial, hipercalcemia, todos de baja incidencia. Dentro de las complicaciones más temidas se encuentra el osteosarcoma visto en los estudios pivotales en ratas, con una dosis suprafisiológicas (X10 veces) y en hueso que siempre modela y crece. A la actualidad y con varios años de experiencia de PTH usada para el tratamiento de osteoporosis solo se reportaron 2 casos de 100.000 pacientes con osteosarcoma, diagnosticados en una paciente que recibió radioterapia en hueso (contraindicación formal) y otra paciente con antecedente de cáncer de mama.
En el año 2016 la FDA aprueba la PTH 1-84 similar a la intacta y que sustituye los efectos fisiológicos de la misma, María Luisa Brandi y colaboradores enuncian las indicaciones:
– Pacientes con manejo inadecuado del calcio que intercurre con enfermedades severas, por falta de compliance, malabsorción, o cualquier intercurrencia que lleve a mayor dosis de sustitución por vía oral.
– Dosis máximas de carbonato de calcio (2.5 grs/día), más de 1.5 ugs/día de vitamina D activa o más de 3 ugrs de análogos de 1α vitamina D.
– Hipercalciuria, litiasis renal o riesgo de, nefrocalcinosis o reducción del clearence de creatinina (menor a 60 ml/min).
– Hiperfosfatemia o producto fosfocálcico mayor a 55 m2/dl2
– Trastorno gastrointestinal con malabsorción.
– Pobre calidad de vida.
La PTH 1-84 presenta mayor vida y con dosis de 50 mcg/día que pueden ir de 25 a 100/día por pQCT aumenta el trabecular y mantiene el cortical.
Se necesitan estudios a largo plazo para evaluar los marcadores de remodelado óseo y su repercusión sobre la masa ósea.
En pediatría en el año 2010 Winner publica un estudio de 12 niños (5 a 14 años) con hipoparatiroidismo crónico, ella no observó trastornos en el crecimiento, los requerimientos de calcio, calcitriol y vitamina D disminuyeron al mínimo, se controló la calcemia y no se evidenció hiperfosfatemia, incluso se normalizaron los niveles de fósforo por estímulo de la fosfaturia. Por DMO se mantuvo el Z excepto en antebrazo que disminuyó.
Como conclusión: el uso de PTH recombinante está aprobado en mayores de 18 años, en menores habría que evaluar cada caso de manera individual y ver riesgo/beneficios de la terapia. Es claro que el beneficio es para aquellos pacientes que presentan altos requerimientos por vía oral, internaciones frecuentes por hipocalcemia sintomática y mala calidad de vida. Las complicaciones crónicas disminuyen, pero aún se necesitan más estudios para evaluar los efectos adversos/beneficios de la terapia a largo plazo.
Dra. Evangelina Giacoia
Medica Especialista en Clínica y Endocrinóloga.
A cargo Área Metabolismo Fosfocálcico. Servicio de Endocrinología, Hospital Nacional Profesor A. Posadas. Docente adscripta a Facultad de Medicina UBA. Secretaria Comisión Directiva Sociedad Argentina de Osteoporosis
Ateneo Clinico ADEPA: Mendoza 2017
Caso clínico: ¿Se puede crecer SIN hormona de crecimiento?
Dra. Sabrina Martin Benítez. Médica Pediatra Endocrinologa, Hospital de Niños P Notti, Mendoza.
Paciente masculino, FN 04/08/06. Antecedentes perinatales: Parto eutócico, Presentación cefálica: EG: 39 semanas, Apgar 7/9, PN 3800 grs., TN: 53 cm. Hipoglucemia a las 24 hs de vida, segundo episodio a los 2 años y 2 meses con convulsión afebril, valor de glucemia confirmado de 21 mg%.
Vagabundeo ocular. Al examen oftalmológico: Hipoplasia de papilas ópticas. Datos clínicos positivos: Depresión del macizo mediofacial. Hipertelorismo, frente amplia. Vagabundeo ocular intermitente. Leve aumento del acúmulo de grasa troncal. Voz aguda.
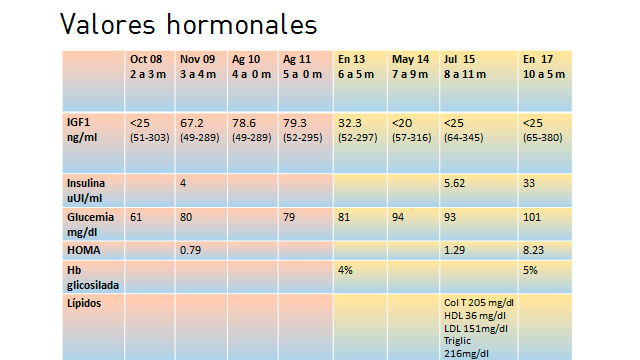
Test de hipoglucemia insulínica para GH (31/10/08): Glucemia -30´: 61mg/dl – GH: 0.71, Basal: glu 50, – GH 1.0, 20´ glu 14 – GH 2.0, 30´ glu 20 – GH 2,5, 60´ glu 61 – GH 1.5. Cortisol basal 7,3 mg/dl 30’: 11 y 60´ 12,1. ACTH 23 pg/ml (h 46), IGF1 <25 ng/ml, IGFBP3: 1,0 ng/ml (0.8 - 3,9), LH 0.34, FSH 1,4, Testosterona <15 ng/dl, PRL 22,2 ng/ml. Prueba compatible con HIPOPITUITARISMO con déficit de GH y ACTH: Se inicia tratamiento con Hidrocortisona 12,5 mg/m2/día. Prueba de GH bajo Arginina (12/12/08): GH basal: 1.8 ng/ml, 30´: 1.1, 45´: 2, 60´: 2.8, y 90´: 1.2: Confirmatoria de déficit absoluto de GH.
En febrero del 2008 inicia tratamiento con Gh luego de otro episodio de hipoglucemia grave, recibiendo corticoides. Se inicia a 0,5 U/kg/sem. RMN de cerebro (04/03/09): Hipófisis pequeña y comprimida a nivel del piso selar con elongación del tallo hipofisario. Aumento de tamaño cisterna supraselar. Defectos a nivel del septum pellucidum
En abril del 2009, presenta avidez por el agua, polidipsia y poliuria. Se realizó test de deprivación hídrica donde se descartó diabetes insípida. En ese momento hizo hipoglucemia a las 21 hs de ayuno (36 mg%). Test de TRH-TSH (22/05/09): Basal: TSH 1.84, 30´: 12.3, 60´: 10.2, 90´: 10.5, 120´: 10.4, y T4L 0.86. Inicia T4 25 ug/día.
Debido a que el paciente presenta velocidad de crecimiento en Pc mayor a 97 en forma sostenida, se disminuye dosis de Gh a 0,27 U/kg/sem y posteriormente el 07/11/11 se suspende para reevaluar. En setiembre del 2012 (luego de 11 meses de suspensión de GH) se reevalúa velocidad de crecimiento 4,1 cm/a.
Se realiza nuevo Test de Arginina del 05/11/12: Basal: 0,13 ng/ml, 15’ 0.19, 30´ 0.27, 60´ 0.30. 08/04/13 se observa que continúa descenso de VC: 6.6 cm/año. Continúa sin tratamiento. 03/04/14 EC: 7 a 8m, VC 4,5 cm/a. Test de arginina: -30´: 0,08, 0´: 0.06, 30´ 0.1, 45´ 0.12, 60´ 0.12, 90´ 0.12. IGF1 <20 (57-316 ng/ml), IGFBP3 3,27 (1,4-6.1 ug/ml).
10/01/14 8a 2m, EO 7,25 años, Dentición atrasada solo 2 incisivos. TSH 2,63, T4L 0,69. Se aumenta dosis de T4 37,5 ug.
22/07/15 Glucemia de 93 mg%, IGF1 < 25 ng/dl, cortisol 2,73 ug/dl, insulina 5,62 mU/ml. Test de Clonidina con priming: -30´ Gh: 0,06, 0´ 0,06, 60´ 0.08, 90´ 0.07, y 120´ 0.07.
05/08/16 RMN de cerebro: Hipoplasia de adenohipófisis con señal homogénea. Hipoplasia del tallo hipofisario y del quiasma óptico. Disminución de tamaño del nervio óptico derecho Neurohipófisis normal.
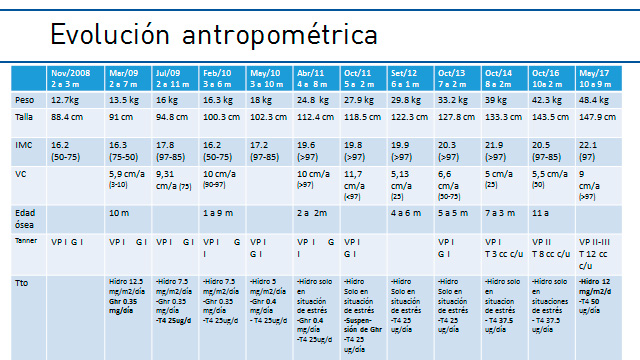
Conclusión: Paciente con displasia septo óptica que inicia tratamiento con GH como emergencia a los 2,62 años por HIPOGLUCEMIA RECURRENTE GRAVE, y déficit absoluto de GH en pruebas confirmatorias, asociado a falta de respuesta de GH a la hipoglucemia espontánea. El tratamiento fue aceptado por Comisión Ampliada debido a que el paciente No presentaba baja talla.
Desde el inicio de GH no repitió hipoglucemias ni convulsiones, con buena progresión del desarrollo neurológico
Durante los dos años de tratamiento iniciales, la talla pasa de Pc 25 a Pc 75-90, encontrándose dentro del máximo de su carril genético. Luego se observa un incremento notable de la velocidad de talla y desviación hacia Pc 97 a pesar de disminuir la dosis a la mitad.
Posteriormente se decide suspensión de tratamiento con Gh y controles estrictos de glucemias permaneciendo el paciente con glucemias siempre por encima de 70 mg% y asintomático.
Luego de un año de suspendido el tratamiento, se reevaluó con Test de Arginina que confirma Déficit absoluto de GH, con pico <1 ng/ml e IGF1 disminuido para la edad.
La velocidad de crecimiento cayó desde 10,49 cm/a hasta 5,12 cm/a, lo que se tradujo en la disminución de talla desde Pc 97 a Pc 90, pero manteniéndose por encima del carril genético. La velocidad de crecimiento intratratamiento era > Pc 97 y post suspensión se encuentra en Pc 10-25.
Como el paciente no ha presentado hipoglucemias post suspensión y su talla actual se encuentra aún por encima del carril genético se decide continuar suspensión del tratamiento.
Última evaluación el día 06/01/16, 10 años 5 meses: Peso: 46 Kg, Talla: 144.9 cm, IMC: 21.9, Tiroides no palpable, Testes 10-12 ml VP 2 incipiente. Analítica: (26/10/16) Hemograma normal. Función renal normal. Glucemia 90 g/dl. Hemoglobina glicosilada 5% Cortisol matinal 5.88 ug/dl. FSH 1.51 U/l, LH 1.17 U/l, Testosterona 39.7 ng/dl (11 h 350), TSH 2.43, T4L 0.88 ug/dl, Prolactina 10.4, GH basal 0.07, IGF1 <25 ng/ml. Rx EO (26/10/16) 11a 3m para EC 10 a 2m. Recibiendo T4 37,5 ug/día. Hidrocortisona solo ante estrés.
Comentario:
La Displasia Septo-óptica (DSO) o Síndrome de Morsier es una entidad rara y altamente heterogénea que se define clínicamente por la combinación de una o más de las siguientes alteraciones: Hipoplasia de nervios ópticos (HNO), malformaciones neuro-radiológicas de línea media (como agenesia de Cuerpo Calloso y agenesia de Septum Pellucidum), e insuficiencia hipofisaria. Ésta última puede presentarse en forma aislada o múltiple. Además, puede estar presente en el período neonatal o desarrollarse progresivamente a lo largo de la vida. Aproximadamente el 30% de las DSO presentan la triada clásica.
La incidencia de esta patología es de 1/10000 recién nacidos vivos. Presenta igual prevalencia en ambos sexos. Se observa más frecuentemente en mujeres jóvenes, primigestas.
En la mayoría de los casos la DSO es una condición esporádica y varias etiologías fueron sugeridas para explicar la patogénesis de la misma como teratógenos ambientales, daño vascular o degenerativo, uso de drogas o alcohol, infecciones virales. Sin embargo, también se han descripto casos familiares. En estos casos mutaciones en factores de transcripción como HESX1, SOX2. SOX3 Y OTX2, fueron implicados en la etiopatogenia. Estos factores son esenciales para el normal desarrollo del cerebro y la glándula pituitaria. Disrupciones en los mismos podrían ser la causa de las características fenotípicas observadas en estos pacientes. La frecuencia de mutaciones halladas en estos genes es muy baja, lo que sugiere que alteraciones en otros genes, probablemente interactuando con el microambiente, contribuyan al desarrollo del síndrome.
Aproximadamente el 30% de los casos de DSO se manifiestan en forma completa. HNO constituye la manifestación clínica más frecuente (75-80%) y es bilateral en el 88% de los casos. Rara vez el compromiso oftalmológico es más severo resultando en microftalmía o anoftalmía.
El compromiso hipofisario es variable, produciendo IGH aislada o múltiple (62%). La deficiencia de hormona de crecimiento constituye la endocrinopatía más frecuentemente hallada, seguida por la deficiencia de la hormona estimuladora de la tiroides (TSH) y hormona adrenocorticotropa (ACTH), mientras que puede no afectarse la secreción de las gonadotropinas. Aunque puede observarse tanto pubertad precoz o fallas en el desarrollo puberal.
El 60% presenta ausencia del septun pellucidum (SP). También se encuentran lesiones en la corteza cerebral, el cuerpo calloso (CC), el sistema olfatorio y el cerebelo. Los déficits neurológicos son comunes, altamente variables y pueden manifestarse desde retardo global del desarrollo hasta déficits focales.
Es difícil predecir cuándo se desarrollará la disfunción hipotálamo hipofisaria (HH). En el trabajo de Cemeroglu y col., dicho compromiso fue diagnosticado antes de los 2 años en la mayoría de los pacientes (86%), por lo tanto, el riesgo de disfunción HH es mayor antes de los 2 años y cuando la HNO y la disgenesia del SP o CC, están presentes. Este riesgo disminuye considerablemente luego de esta edad. En ningún paciente el diagnosticado se realizó luego de los 8 años. La DBT insípida se presentó exclusivamente antes de los 4 años. Por lo que este grupo propone realizar controles de función hipofisaria cada 4 ─ 6 meses los primeros 2 años, cada 6 ─ 12 meses entre los 2 y 8 años y luego anualmente durante la pubertad.
Otros hallazgos menos frecuentemente observados en los pacientes con DSO son:
Trastorno del sueño: En los núcleos supra-quiasmáticos del hipotálamo anterior se localiza el reloj biológico. Estos núcleos se ubican por encima del quiasma óptico y reciben información de los NO para sincronizar el reloj biológico al ciclo de 24 hs Luz-oscuridad. Es necesario re-setear el ritmo circadiano diariamente a través del estímulo visual, por lo que las alteraciones en la visión pueden interferir con el sistema circadiano y así producir efectos perniciosos sobre el comportamiento.
Sobrepeso/obesidad por alteración del núcleo ventro-medial del tálamo que suprime el hambre y la ingesta en respuesta a la leptina.
Retardo del desarrollo neurológico, el que es altamente variable en su manifestación clínica.
Los pacientes con DSO y deficiencia de hormona de crecimiento (HC) se benefician con el tratamiento con dicha hormona; sin embargo, se ha descripto también un subgrupo de pacientes que a pesar de presentar deficiencia de HC presentan una talla adulta normal. Se ha sugerido que niveles elevados de insulina, prolactina o leptina podrían promover el crecimiento independiente de HC, aunque el mecanismo aún no está completamente dilucidado.
El paciente que se discutió en este encuentro presentaba deficiencia multihormonal (HC, ACTH y TSH) en el contexto de una DSO, por lo que se sustituyó adecuadamente con levotiroxina, hidrocortisona y hormona de crecimiento. Debido a que durante el tratamiento con HC el paciente crece con velocidad de crecimiento mayor al percentilo 97, se decide suspender dicho tratamiento. A pesar de la suspensión de la terapia con HC el paciente continúa creciendo adecuadamente, en rango genético.
Dra. Maria Isabel Di Palma. Medica Pediatra Endocrinóloga. Hospital Nacional de Pediatría JP Garrahan, CABA.